1. Why did I receive two files per sample named R1 and R2?
If the sequencing configuration of your project is paired-end, you will get two reads per fragment with the first read in forward orientation, and the second read in reverse-complement orientation. This generates a read pair – R1 and R2 – that together describe a nucleic acid molecule as sequenced from either end in a 5’->3’ direction (anti-parallel). Corresponding reads from the R1 and R2 files are collectively a single paired-end read.
For a visual representation of the same, please examine the following image published by Illumina®:
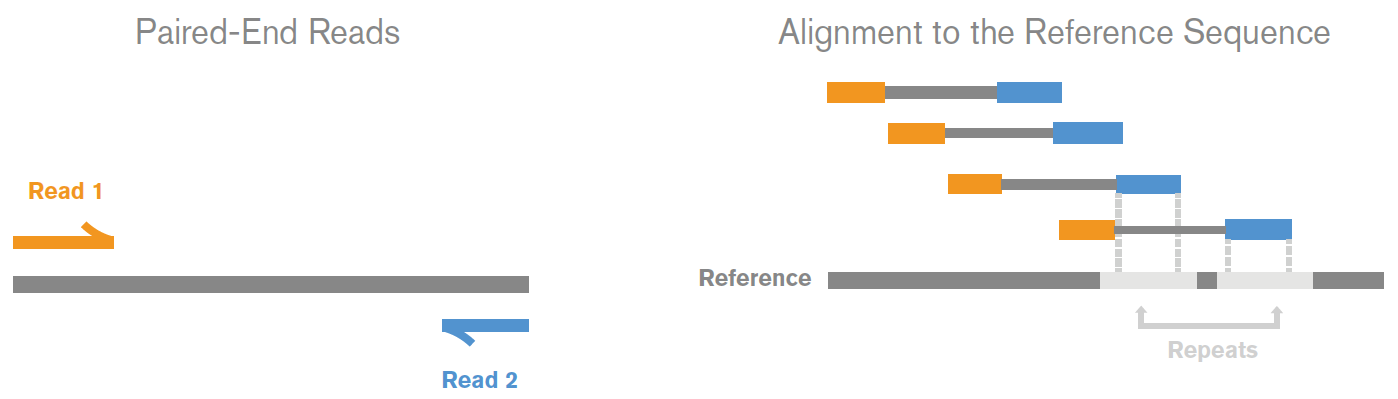
Paired-end sequencing enables both ends of the DNA fragment to be sequenced. Because the distance between each paired read is known, alignment algorithms can use this information to map the reads over repetitive regions more precisely. This results in much better alignment of the reads, especially across difficult-to-sequence, repetitive regions of the genome. Source: Illumina
If a 10x Genomics® library was sequenced, you may receive additional files such as I1 and I2, which contain further information necessary for downstream Cell Ranger applications.
2. Why is my sample name different from the delivered file name?
The Illumina bcl2fastq demultiplexing tool only accepts sample sheets with letters, numbers, and dashes (-). Any other symbol will need to be converted to those for the program to run.
3. In general, will I receive high-quality sequencing data?
The “% Base >= 30” refers to the number of bases that had a Phred quality score greater than 30. The Phred quality score is a logarithmic scale used to measure the accuracy of base calls based on sequencer-defined probability of error in each base call.
Illumina provides sequencing quality scores that measure the probability that a base is called incorrectly. In our provided report, the “% Bases >=30” indicates the percentage of bases that have a sequencing accuracy of 99.9%. Details about the quality score can be found here: https://www.illumina.com/science/technology/next-generation-sequencing/plan-experiments/quality-scores.html.
Illumina has recommended quality parameters for different configurations/platforms, which you can find below.
MiSeq™: https://www.illumina.com/systems/sequencing-platforms/miseq/specifications.html
HiSeq® 3000/4000: https://www.illumina.com/systems/sequencing-platforms/hiseq-3000-4000/specifications.html
NovaSeq™: https://www.illumina.com/systems/sequencing-platforms/novaseq/specifications.html
4. Are the reads I got adapter-trimmed already?
If your project was performed on the HiSeq or NovaSeq platform, then no, our delivered reads have not been trimmed. We recommend you use trimming tools to remove low-quality and adapter sequences from the data.
If your project was performed on the MiSeq platform, then yes, our delivered reads are adapter-trimmed already.
5. Do you deliver BCL files?
Due to the number of files and size of BCL files, we do not provide these by default. We may be able to provide these by request for an additional cost.
6. What are our options for data delivery?
We offer different options for data delivery, including hard drive, sFTP, and cloud delivery via AWS, Google Cloud, etc. Additional charges may apply for hard drive and sFTP delivery depending on the amount of data being delivered.
Have a specific question?
Contact Us | Phone 1-877-GENEWIZ (436-3949), ext. 1